A Pipeline for Functionally Annotating Pangenomes
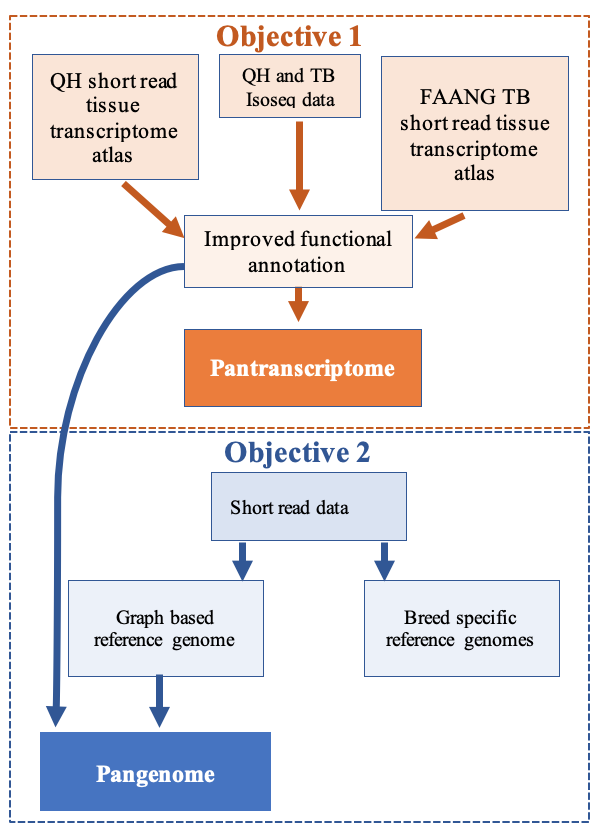
How an individual’s genetic background influences different traits - for example, coat color, disease risk, etc. - is a fundamental question for the genomics community. Increasingly affordable technology has made identification of genetic markers that contribute to many disease and performance traits commonplace. However, identifying the markers that are functional is challenging, especially for complex traits where multiple markers contribute to the trait. Currently, we rely on the “reference” genome from a single individual to identify the markers that are predicted to be functional for each trait. However, there are numerous limitations of using a single reference genome, including: 1) a single individual does not represent all the genetic variation that is normal within individuals and between breeds and populations; 2) the individual reference genome may contain missing sequence that is present in distantly related individuals; and 3) does not represent larger structural variants that are present in normal individuals. These limitations mean that variation not represented by the reference genome are missed when identifying variants that contribute to traits. This is particularly problematic when using individuals that are distantly related to the reference genome individual. Many of these limitations can be overcome by creating a “pangenome.” The pangenome incorporates genomic information from multiple individuals leading to a reference genome that is more representative of the genetic background of a species rather than just a single individual.
Two MSI PIs from the College of Veterinary Medicine, Professor and Associate Dean for Research Molly McCue and Assistant Professor Sian Durward-Akhurst, are working on a project called “A robust pipeline to functionally annotate non-traditional model pangenomes.” They seek to facilitate genome research efforts in domestic animals by providing tools and readily accessible data that will expedite efforts to accurately identify underlying genetic causes for traits of interest including disease (e.g.,risk of muscle disease and cardiac arrhythmias) and performance (e.g., growth rates in livestock) in relevant, non-traditional animal models. This project capitalizes on their already curated large datasets of short-read genomes (> 1,000) and gene expression data (>30 tissues) in the domestic horse. They will develop further resources to accelerate understanding of the link between the genomeand traits of interest by building scientific protocols and computational tools to create the first equine pangenome. These tools can then be applied to any species with available species-specific data. This will also improve our understanding of where genes and other critical genome elements are located in the genome sequence. This project recently received a Research Computing Seed Grant.
As of September 2023, the RC Seed Grant programs have been revised into the DSI Seed Grant programs. DSI Seed Grants include many of the same goals as the old program, with a new emphasis on data science. Complete information about DSI Seed Grants, including application deadlines, can be found on the RC website.